#RAD PROTOCOL
>Olympia Oyster - **Initial outplant**
>32 samples from three populations
>Layout:
_use filter tip pipette tips at all times, to avoid cross-contamination between libraries_
* Extract DNA
> Samples taken from field August 2013
> 
> [more details](http://olyo.wikispaces.com/August+2013+outplanting)
>
> Qiagen 96 well plate used for DNA extraction
>
> Tissue into dry plate - then buffer added - [details](http://genefish.wikispaces.com/Sam%27s+Working+Notebook+August+-+December+2014#sjw20141014)
>
* Check Quantity and Quality
> Gel (representative pic)
> 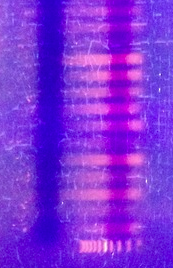
> complete gels pics and details
> [1](http://sr320.tumblr.com/post/100190074759)
> [2](http://sr320.tumblr.com/post/100231194034)
> [3](http://sr320.tumblr.com/post/100245499294)
>
> _side note_
> later used DNAzol with [slightly better results](http://sr320.tumblr.com/post/101186109739) with respect to HMW DNA
>
> DNA concentrations
> [](https://docs.google.com/spreadsheets/d/1ikRj2DEvkgIyogSGpl2V_6a64XDUyCSlwdQFO8Qv1no/edit#gid=1025947309)
> [details](http://genefish.wikispaces.com/Sam%27s+Working+Notebook+August+-+December+2014#sjw20141022)
* Transfer **500 ng** of genomic DNA from each sample to a PCR plate.
* Air dry the DNA overnight.
>
> Samples required two days of drying.
> In hindsight - _probably could have pushed forward after overnight._
>
* Resuspend DNA in a total volume of 20 µL of water.
* Make restriction enzyme digestion mix
Reageant | amount
------------ | -------------
H2O | 2.2 µL
CutSmart Buffer (10X) | 2.5 µL
SbfI-HF (NEB R3642L) | 0.25 µL
RNase A (100 mg/mL) | 0.05 µL
Add 5µL to each sample.
_note that PstI will also work with adaptors_
* Incubate at 37˚C for 90 minutes, then at 80˚C for 20 minutes to inactivate the enzyme (_check appropriate inactivation temperature for the enzyme in use, if other than SbfI_).
* Add 2 µL of each barcoded SbfI short P1 RAD adapter (25nM) (these are the illumina barcodes located in -20˚C freezer) to each digested sample. Mix by pipetting. Use RAD adapters 10 times from each aliquot plate before throwing away.
*
>
>Added 2uL of each adapter to corresponding well of SbfI digested DNA (e.g. DNA plate well A1 got the P1 adapter from >well A1 in the adapter plate).
>
_Note: now is a good time to turn on the sonicator to bring it down to temperature._
* Make ligation mix:
Reageant | amount
------------ | -------------
H2O | 1.95µL
NEBuffer 2 (10X) | 0.5µL
rATP (100mM Fermentas R0441) | 0.3µL
T4 DNA Ligase (NEB M0202M) | 0.25µL
Add 3 µL to each sample.
* Incubate the plate at 20˚C for 60 minutes, then at 65˚C for 30 minutes to inactivate the enzyme.
* Combine the samples that are to be sequenced together into libraries. Combine samples with lower quality DNA into the same pool to reduce PCR bias at the end of the library prep. The number of samples to combine into each library is dependent on DNA concentration, genome size and sequencer capacity. For hard to replace DNAs, reserve ½ of library in case there is problem downstream. To combine 12 samples from a library: 60 µL / sample x 12 samples = 720 µL. Reserve 20 µL aside before sonication to allow running un-sonicated sample on the gel (with the sonicated sample that is run in step 7). Split the remaining 700 µL into two aliquots of 350 µL in 1.5 mL tubes for sonication. NOTE: This is a good time to put EB in the incubator (55˚C) for today’s Minelute purification steps.
* Sonicate the libraries using the QSonica sonicator. Make sure that the water level is filled to the level indicated in the water batch. Turn on the water bath at least an hour before sonication to allow it to cool to 4˚C. Turn on the sonicator and set to 25% power, 4 cycles of 30 s on, 59 s off (will read 2:00 sonication). Load the samples into the wheel, making sure that the unused slots contain 1.5 mL vials with the same amount of liquid. Samples all need to be about 350 ul for the NWFSC standard sonication method. After sonication, combine the two pools from the same library together into a 5 mL tube to allow enough volume for Minelute cleanup.
>
>Created two separate pools of "low" and "high" quality for shearing.
>Low quality samples (5uL from each):
>All rows, columns 1 -9
>
>Higher quality samples (5uL from each):
>All rows, columns 10 -12
>
>Sheared each samples with the following cycling protocols on the Biorupter Plus (Diagenode):
>
>Low
>3 cycles of:
>30 seconds on
>59 seconds off
>
>High
>4 cycles of:
>30 seconds on
>59 seconds off
>
* Run a test gel of sonicated and un-sonicated product using a 1% Gel Red gel (50 mL 1X TAE with 2.5 uL GelRed). Load 10 uL each of the sonicated and unsonicated product to evaluate whether the desired size range is present. If sonication looks good, combine the two sonication pools from the same library together in a single 5 mL conical tube and proceed with the Qiagen PCR Purification in the next step.
>
>Ran a subset of sheared gDNA (5uL from each pool) on gel to verify final size range:
>
> >
>
>Gel loading:
>
>Lane 1 - O'GeneRuler 100bp Ladder (ThermoFisher)
>
>Lane 2 - Low quality
>
>Lane 3 - Higher quality
>
>
>
>I neglected to run a set of un-sheared DNA.
>Both samples appear to have an average size of 200 - 400bp.
>
>
>
>After confirming satisfactory shearing, the two samples were combined and run on a 1% agarose low TAE gel (stained >with EtBr) for size selection.
>
>
>O'GeneRuler 100bp Ladder (ThermoFisher)
>
>
>
>
>
>Gel loading:
>
>Lane 1 - O'GeneRuler 100bp Ladder (ThermoFisher)
>
>Lane 2 - Low quality
>
>Lane 3 - Higher quality
>
>
>
>I neglected to run a set of un-sheared DNA.
>Both samples appear to have an average size of 200 - 400bp.
>
>
>
>After confirming satisfactory shearing, the two samples were combined and run on a 1% agarose low TAE gel (stained >with EtBr) for size selection.
>
>
>O'GeneRuler 100bp Ladder (ThermoFisher)
>
>
>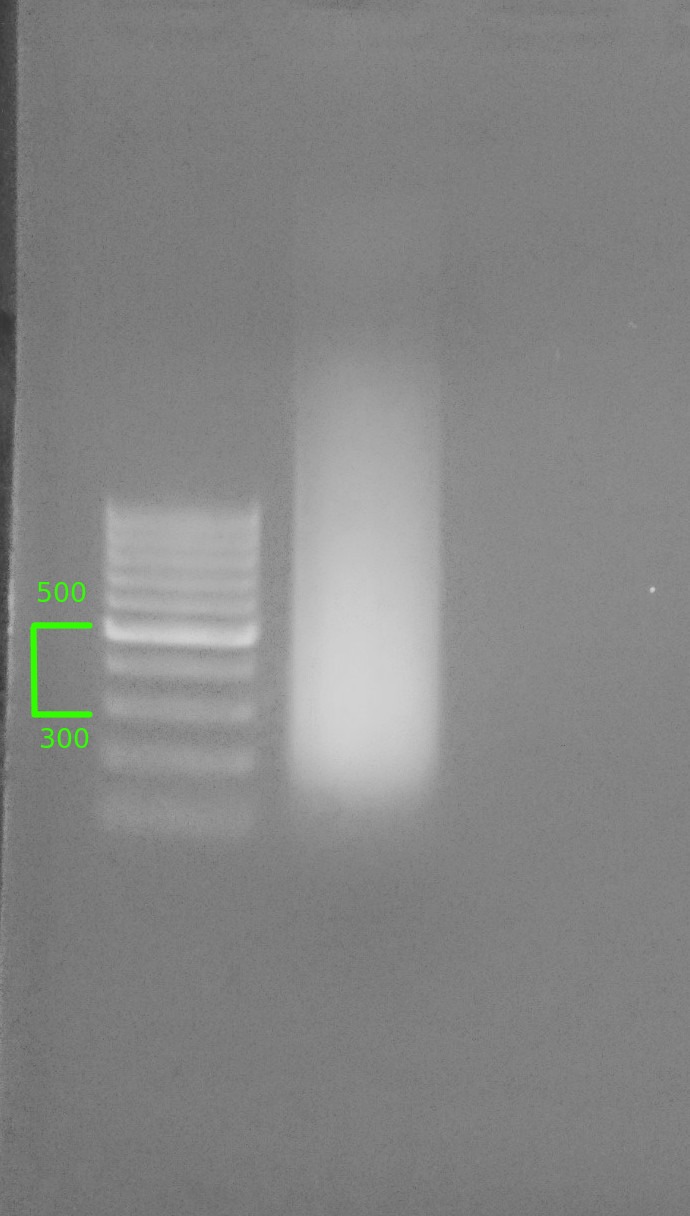 >
>
>
>
>
>Size range of sheared DNA from 300 - 500bp was excised from gel.
>
>
>
>O'GeneRuler 100bp Ladder (ThermoFisher)
>
>
>
>
>
>
>
>
>Size range of sheared DNA from 300 - 500bp was excised from gel.
>
>
>
>O'GeneRuler 100bp Ladder (ThermoFisher)
>
>
> >
>
>
* Purify the sample with the Qiagen MinElute PCR Purification Kit. Elute with 15µL EB.
* Gel extract the sample. Run the entire sample on exactly 1% agarose gel (100 – 120 V from 30 – 45 min) and use 100 bp DNA Ladder as a reference to extract the proper size range. It’s easiest to use SybrSafe gel stain at this step, and the dark reader to visualize the gel for cutting the band. Add 5 uL SybrSafe to agarose before pouring the gel. The dark reader, with an orange filter, can be used to visualize the gel, and the old Polaroid to take a photo. Use a clean razor blade to extract the smear between 400-600 bp. Place the cut gel plug in clean micro tube and weigh gel plug so that you know what the ‘volume’ is for Minelute cleanup.
>
>Gel fragment weighed 254mg.
>
>Purified using MiniElute Gel Extraction Kit (Qiagen).
>Added three volumes (762uL) of Buffer QG to gel slice.
>Incubated ~10mins on rotator until gel slice was fully dissolved.
>Added one gel slice volume (254uL) of isopropanol; inverted multiple times to mix.
>Added 700uL to MiniElute column; spun max speed (~16,000g) 1min; discarded flow-through.
>Added remainder of sample to MiniElute column; spun max speed (~16,000g) 1min; discarded flow-through.
>Added 500uL of Buffer QG to MiniElute column; spun max speed (~16,000g) 1min; discarded flow-through.
>Added 750uL of Buffer PE to MiniElute column; incubated @ RT for 5mins; spun max speed (~16,000g) 1min; discarded >flow-through.
>Spun MinElute column spun max speed (~16,000g) 1min; transferred column to clean 2.0mL tube.
>Added 50uL of Buffer EB to column, incubated @ RT for 5mins and spun max speed (~16,000g) 1min; discarded column.
>
>Stored sample @ 4C.
>
>
---
**LOTS OF STUFF**
---
* Make PCR mix. The amount of purified DNA can vary in the PCR. Sample pools with lower quality DNA may require more DNA in this PCR step. Each PCR volume must be 100µL total so adjust the amount of water you use.
Reagent | amount
------------ | -------------
H2O | 36µL (can be variable)
2X Phusion Master Mix (NEB F-531L) | 50 µL
RAD P1 primer (10µM) | 2 µL
RAD P2 primer (10µM) | 2 µL
Purified sample from step 15 (Store remaining at -20˚C | 10 µL (can be variable)
| 100µl
Place in thermal cycler and perform PCR. Cycling conditions are 98˚C for 30 sec; 14X {98˚C for 10 sec, 65˚C for 30 sec, 72˚C for 30 sec}; 72˚C for 5 minutes; hold at 10˚C. NOTE: fewer (as few as 12 cycles) cycles of PCR are better to avoid PCR artifacts in the libraries.
* Purify the sample with Qiagen MinElute PCR Purification Kit. Elute with 20µL EB.
* Gel extract the sample to remove the low molecular weight adapter and primers. Run on 1% agarose gel with SybrSafe and 100 bp DNA Ladder as a reference and extract the 400-600 bp band. Place gel plug into a clean micro tube and weigh gel plug.
* Use Qiagen Gel Purification in the Minelute columns (following instructions for extra steps for products that will be used for sequencing, as above). Elute with 14.5 µL EB. Then add 1.5µL of EB containing 1% Tween-20.
*Run qPCR (KAPA kit) and Qbit the samples. Dilute samples for submission to sequencing facility.
>
>
>
* Purify the sample with the Qiagen MinElute PCR Purification Kit. Elute with 15µL EB.
* Gel extract the sample. Run the entire sample on exactly 1% agarose gel (100 – 120 V from 30 – 45 min) and use 100 bp DNA Ladder as a reference to extract the proper size range. It’s easiest to use SybrSafe gel stain at this step, and the dark reader to visualize the gel for cutting the band. Add 5 uL SybrSafe to agarose before pouring the gel. The dark reader, with an orange filter, can be used to visualize the gel, and the old Polaroid to take a photo. Use a clean razor blade to extract the smear between 400-600 bp. Place the cut gel plug in clean micro tube and weigh gel plug so that you know what the ‘volume’ is for Minelute cleanup.
>
>Gel fragment weighed 254mg.
>
>Purified using MiniElute Gel Extraction Kit (Qiagen).
>Added three volumes (762uL) of Buffer QG to gel slice.
>Incubated ~10mins on rotator until gel slice was fully dissolved.
>Added one gel slice volume (254uL) of isopropanol; inverted multiple times to mix.
>Added 700uL to MiniElute column; spun max speed (~16,000g) 1min; discarded flow-through.
>Added remainder of sample to MiniElute column; spun max speed (~16,000g) 1min; discarded flow-through.
>Added 500uL of Buffer QG to MiniElute column; spun max speed (~16,000g) 1min; discarded flow-through.
>Added 750uL of Buffer PE to MiniElute column; incubated @ RT for 5mins; spun max speed (~16,000g) 1min; discarded >flow-through.
>Spun MinElute column spun max speed (~16,000g) 1min; transferred column to clean 2.0mL tube.
>Added 50uL of Buffer EB to column, incubated @ RT for 5mins and spun max speed (~16,000g) 1min; discarded column.
>
>Stored sample @ 4C.
>
>
---
**LOTS OF STUFF**
---
* Make PCR mix. The amount of purified DNA can vary in the PCR. Sample pools with lower quality DNA may require more DNA in this PCR step. Each PCR volume must be 100µL total so adjust the amount of water you use.
Reagent | amount
------------ | -------------
H2O | 36µL (can be variable)
2X Phusion Master Mix (NEB F-531L) | 50 µL
RAD P1 primer (10µM) | 2 µL
RAD P2 primer (10µM) | 2 µL
Purified sample from step 15 (Store remaining at -20˚C | 10 µL (can be variable)
| 100µl
Place in thermal cycler and perform PCR. Cycling conditions are 98˚C for 30 sec; 14X {98˚C for 10 sec, 65˚C for 30 sec, 72˚C for 30 sec}; 72˚C for 5 minutes; hold at 10˚C. NOTE: fewer (as few as 12 cycles) cycles of PCR are better to avoid PCR artifacts in the libraries.
* Purify the sample with Qiagen MinElute PCR Purification Kit. Elute with 20µL EB.
* Gel extract the sample to remove the low molecular weight adapter and primers. Run on 1% agarose gel with SybrSafe and 100 bp DNA Ladder as a reference and extract the 400-600 bp band. Place gel plug into a clean micro tube and weigh gel plug.
* Use Qiagen Gel Purification in the Minelute columns (following instructions for extra steps for products that will be used for sequencing, as above). Elute with 14.5 µL EB. Then add 1.5µL of EB containing 1% Tween-20.
*Run qPCR (KAPA kit) and Qbit the samples. Dilute samples for submission to sequencing facility.