#methods-RNAseq
Total RNA was extracted using Tri-Reagent per manufacturer’s instruction. Potential DNA carry-over was removed from extracted RNA using the Turbo DNA-free treatment according to the manufacturer's instructions (Ambion). Messenger RNA (mRNA) enrichment, library preparation, and sequencing was performed by GENEWIZ. Poly(A) enrichment was performed using NEBNext® Poly(A) mRNA Magnetic Isolation Module (New England Biolabs). Illumina sequencing libraries were prepared with the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (New England Biolabs) and each library was uniquely barcoded with the NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1) (New England Biolabs). All six libraries were multiplexed and run on a single lane on a HiSeq2500 (Illumina), with a read length of 100bp (single end).
RNA-seq analysis was carried out using the TopHat2 suite in the iPlant Collaborative Discovery Environment. Specifically TopHat2-SE was used to align the six libraries (3 individuals, pre and post heat shock) to the _Crassostrea gigas_ genome.
Below find the specific procedure presented in a tutorial-based method.
---
####Task 1: Align read data to _Crassostrea gigas_ genome.
Tophat is a specialized alignment software for RNA-seq reads that is aware of splice junctions when aligning to a reference assembly.
1) Click Apps from DE workspace and select **TopHat2-SE**. Use search bar.
 2) Under 'Analysis Name' leave as defaults or make desired changes.
3) Under **Input data** for FASTQ files add six fastq.gz files located in `paper-Cg-temp-methylation/raw/` with prefixes _2M, 2M-HS, 4M, 4M-HS, 6M, 6M-HS_.
4) Under **Reference Genome** for 'Provide a reference genome file in FASTA format' select `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.dna_sm.toplevel.fa`
5) For **Reference Annotations** add the GTF file `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.gtf`
6) Click **Launch Analyses** and monitor the status of you job (this takes ~10 hours)
---
####Task 2: Assemble transcripts using **Cufflinks2**
Cufflinks assembles RNA-Seq alignments into a parsimonious set of transcripts, then estimates the relative abundances of these transcripts based on how many reads support each one, taking into account biases in library preparation protocols. A detailed manual can be found at http://cole-trapnell-lab.github.io/cufflinks/manual/.
1) Open **Cufflinks2**
2) For **Input Data** add the six bam files from the `bam` subdirectory of the TopHat2 output (`/paper-Cg-temp-methylation/analyses/TopHat2-SE_analysis_heat-b-2014-12-19-15-09-00.4/bam/`)
3) Under **Reference Sequence** use custom option select `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.dna_sm.toplevel.fa`
4) For **Reference Annoations** add the GTF file `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.gtf`
5) Click **Launch Analyses** and monitor the status of you job (This takes about 2 hours).
---
####Task 3: Merge all Cufflinks transcripts into a single transcriptome annotation file using **Cuffmerge2**
Cuffmerge merges together several Cufflinks assemblies. It handles also handles running Cuffcompare. The main purpose of this application is to make it easier to make an assembly GTF file suitable for use with Cuffdiff. A merged, empirical annotation file will be more accurate than using the standard reference annotation, as the expression of rare or novel genes and alternative splicing isoforms seen in this experiment will be better reflected in the empirical transcriptome assemblies.
1) Open the **Cuffmerge2** app. Under 'Input Data', browse to the results of the Cufflinks analyses (above) and add the 6 gtf files located in the `gtf` subdirectory (`/paper-Cg-temp-methylation/analyses/Cufflinks2_analysis_heat-2014-12-20-15-57-48.9/gtf/`).
2) For **Reference Annotations** add the GTF file `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.gtf`
3) Under **Reference Sequence** use custom option select `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.dna_sm.toplevel.fa`
4) Click **Launch Analyses** and monitor the status of you job.
---
####Task 4: Compare expression analysis using CuffDiff2
Cuffdiff is a program that uses the Cufflinks transcript quantification engine to calculate gene and transcript expression levels in more than one condition and test them for significant differences.
1) Open **Cuffdiff2**. For 'Input Data' Sample 1 Name enter _Pre_ and add three bam file from Tophat analysis (`paper-Cg-temp-methylation/analyses/TopHat2-SE_analysis_heat-b-2014-12-19-15-09-00.4/bam/`).
2) Under 'Analysis Name' leave as defaults or make desired changes.
3) Under **Input data** for FASTQ files add six fastq.gz files located in `paper-Cg-temp-methylation/raw/` with prefixes _2M, 2M-HS, 4M, 4M-HS, 6M, 6M-HS_.
4) Under **Reference Genome** for 'Provide a reference genome file in FASTA format' select `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.dna_sm.toplevel.fa`
5) For **Reference Annotations** add the GTF file `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.gtf`
6) Click **Launch Analyses** and monitor the status of you job (this takes ~10 hours)
---
####Task 2: Assemble transcripts using **Cufflinks2**
Cufflinks assembles RNA-Seq alignments into a parsimonious set of transcripts, then estimates the relative abundances of these transcripts based on how many reads support each one, taking into account biases in library preparation protocols. A detailed manual can be found at http://cole-trapnell-lab.github.io/cufflinks/manual/.
1) Open **Cufflinks2**
2) For **Input Data** add the six bam files from the `bam` subdirectory of the TopHat2 output (`/paper-Cg-temp-methylation/analyses/TopHat2-SE_analysis_heat-b-2014-12-19-15-09-00.4/bam/`)
3) Under **Reference Sequence** use custom option select `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.dna_sm.toplevel.fa`
4) For **Reference Annoations** add the GTF file `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.gtf`
5) Click **Launch Analyses** and monitor the status of you job (This takes about 2 hours).
---
####Task 3: Merge all Cufflinks transcripts into a single transcriptome annotation file using **Cuffmerge2**
Cuffmerge merges together several Cufflinks assemblies. It handles also handles running Cuffcompare. The main purpose of this application is to make it easier to make an assembly GTF file suitable for use with Cuffdiff. A merged, empirical annotation file will be more accurate than using the standard reference annotation, as the expression of rare or novel genes and alternative splicing isoforms seen in this experiment will be better reflected in the empirical transcriptome assemblies.
1) Open the **Cuffmerge2** app. Under 'Input Data', browse to the results of the Cufflinks analyses (above) and add the 6 gtf files located in the `gtf` subdirectory (`/paper-Cg-temp-methylation/analyses/Cufflinks2_analysis_heat-2014-12-20-15-57-48.9/gtf/`).
2) For **Reference Annotations** add the GTF file `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.gtf`
3) Under **Reference Sequence** use custom option select `paper-Cg-temp-methylation/genome/Crassostrea_gigas.GCA_000297895.1.24.dna_sm.toplevel.fa`
4) Click **Launch Analyses** and monitor the status of you job.
---
####Task 4: Compare expression analysis using CuffDiff2
Cuffdiff is a program that uses the Cufflinks transcript quantification engine to calculate gene and transcript expression levels in more than one condition and test them for significant differences.
1) Open **Cuffdiff2**. For 'Input Data' Sample 1 Name enter _Pre_ and add three bam file from Tophat analysis (`paper-Cg-temp-methylation/analyses/TopHat2-SE_analysis_heat-b-2014-12-19-15-09-00.4/bam/`).
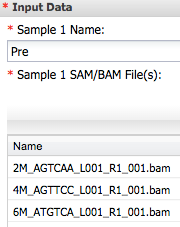 For Sample 2 enter _Post_ and add other three bam files ...
For Sample 2 enter _Post_ and add other three bam files ...
 2) Next, open the **Reference Annotations** section and add a custom reference annotation file using the `merged_with_ref_ids.gtf` file from the Cuffmerge output folder (`paper-Cg-temp-methylation/analyses/Cuffmerge2_heat-2014-12-20-19-14-34.8/cuffmerge_out/merged_with_ref_ids.gtf`).
3) Click **Launch Analyses** and monitor the status of you job.
4) Upon completion several outputs will be produced (see Results section).
directory: `cuffdiff_out`
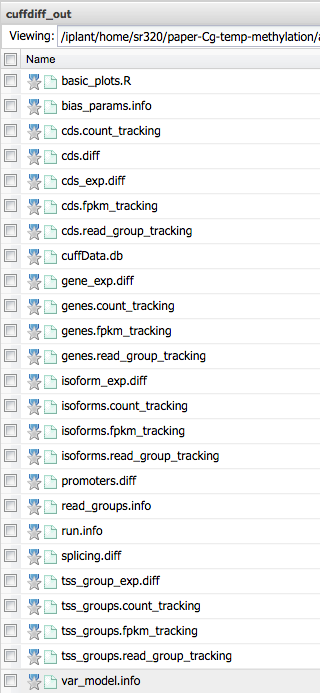
directory: `graphs`
directory: `sorted_data`
2) Next, open the **Reference Annotations** section and add a custom reference annotation file using the `merged_with_ref_ids.gtf` file from the Cuffmerge output folder (`paper-Cg-temp-methylation/analyses/Cuffmerge2_heat-2014-12-20-19-14-34.8/cuffmerge_out/merged_with_ref_ids.gtf`).
3) Click **Launch Analyses** and monitor the status of you job.
4) Upon completion several outputs will be produced (see Results section).
directory: `cuffdiff_out`

directory: `graphs`

directory: `sorted_data`
