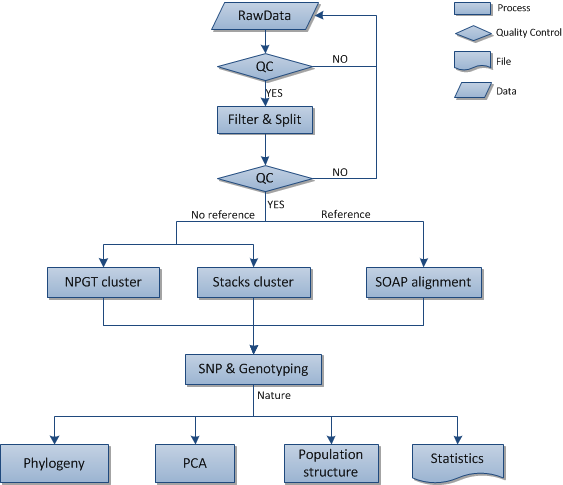
测序得到的原始图像数据经base calling转化为fastq格式的原始下机序列数据 (raw data或raw reads),fastq格式文件内容如下所示:
@FCB068CABXX:6:1101:1403:2159#TAGGTTAT/1 GTAGAAGACTTATAGATTAAAATTCTCCAACATATAGATGTCCTTACACCGTTTTCCTTTGCTCAGCAGGCTCCGTGTTTGCTTGTCCTT + c`bcc_c^ccde_df\c_aeff`ffcfffdfedadca^`b_eed`fe\fed\babdba^Yeebeccfdeae_eec^dbXbda`]bcbebc
在fastq文件中每个read共有4行信息,第1行和第3行是序列名称(通常为了节省存储空间,第三行“+”后面的序列名称会被省略),由测序仪产生;第2行是序列;第4行是序列的测序质量,与第2行各碱基一一对应。用第四行每个字符对应的ASCII值减去64,即得到该碱基的测序质量值。例如,c对应的ASCII值为99,那么其对应的碱基质量值是35。表1为测序错误率与测序质量值简明对应关系。具体地,如果测序错误率用E表示,碱基质量值用sQ表示,则有下列关系:
| 测序错误率 | 测序质量值 | 对应字符 |
|---|---|---|
| 5% | 13 | M |
| 1% | 20 | T |
| 0.1% | 30 | ^ |
对于有参考基因组的物种,所有测序所得的reads经过SOAPaligner[1](http://soap.genomics.org.cn)与参考序列进行比对。在比对时,所有reads都经过了如下的处理:如果一个原始的read无法比对上参考序列,其第一个和最后两个碱基将会被去除,之后再和参考序列进行比对。如果仍然无法匹配,则再去掉最后两个碱基再比对,如此迭代进行,直到能够匹配或reads短于40bp为止。如果reads短于40bp仍无法匹配,则丢弃。然后根据比对结果,计算出平均深度和覆盖度。
对于无参考基因组的物种,我们使用Stacks[2](http://creskolab.uoregon.edu/stacks/)或华大自主开发的NPGT软件包对read1进行聚类,并依据聚类结果进行下一步的snp检测。
对于有参考基因组的物种,根据比对结果,我们使用soapsnp[3]软件综合考虑数据特征、测序质量及实验方面的影响因素,利用贝叶斯模型,在实际观察到的数据基础上计算出每个可能的基因型概率。挑选出概率最大的基因型作为该测序个体在特定位点的基因型,在此基础上给出一个反映该基因型准确度的质量值,并得到一致序列(一组含有位点比对信息的参考序列)。在一致序列的基础上,对于参考序列上存在着多态性的位点进行筛选和过滤,符合以下四个条件的SNP位点将予以保留:
对于无参考基因组的物种,我们使用NPGT软件包中的radsnp,通过聚类来进行read1上的SNP检测。
整合各个样品的SNP检测结果,过滤掉在样品中缺失率较大(默认大于等于50%)的SNP位点,生成分型列表。
系统进化树(phylogenetic tree,又称evolutionary tree进化树)就是描述群体间进化顺序的分支图或树,表示群体间的进化关系。进行SNP检测之后,得到的个体SNPs可以用于计算种群之间的距离。两个体i和j之间的p-距离通过如下公式计算:
公式中L为高质量SNPs区域长度,在位置1的等位基因为A/C,那么
| dij(l)=0 | 如果两个个体的基因型是AA和AA |
| dij(l)=0.5 | 如果两个个体的基因型是AA和AC |
| dij(l)=0.5 | 如果两个个体的基因型是AC和AC |
| dij(l)=1 | 如果两个个体的基因型是AA和CC |
运用MEGA5.0[4](http://mega.software.informer.com/5.0/)或TreeBeST(http://treesoft.sourceforge.net/treebest.shtml)或PHYLIP(http://evolution.genetics.washington.edu/phylip.html)软件计算距离矩阵,以此为基础,通过邻接法(neighbor-joining method)构建系统进化树,重复计算引导值(bootstrap values)达1000次,计算基于所有的SNPs。
主成分分析(Principal Component Analysis, PCA)是指通过线性变换找出数据集中对方差贡献最大的主要成分,以降低数据复杂度的一种分析。为减少数据干扰,此分析仅用于常染色体SNP数据,且忽略出现多余2个等位基因的位点以及错配数据。
在个体i中,k位点的SNP用dik表示,若个体i与参考等位基因是纯合,dik=0;如果是杂合,dik=1;如果个体i与非参考等位基因是纯合,dik=2。M是一个包含标准基因型的n ×S的矩阵:
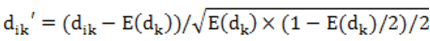
公式中E(dik)是dk的平均值,个体样本协方差n×n矩阵通过X=MMT/S进行计算。X的特征向量分解应用功能特征函数R完成。我们使用EIGENSOFT[5]软件提供的twstats 程序进行Tracey-Widom 检验得到特征向量的显著性分析。
群体遗传结构指遗传变异在物种或群体中的一种非随机分布。按照地理分布或其他标准可将一个群体分为若干亚群,处于同一亚群内的个体亲缘关系较高,而亚群与亚群之间则亲缘关系稍远。群体结构分析有助于理解进化过程,并且可以通过基因型和表型的关联研究确定个体所属的亚群。FRAPPE[6]应用最大似然法来推断在假设个体来自于K个祖先群的情况下,每个个体的遗传祖先。
upload
|---- Cleandata cleanreads文件夹
| |--- A.fq.gz A样品cleanreads
| |--- B.fq.gz B样品cleanreads
|---- Data.stat.xls 所有样品数据统计列表
|---- SNP.stat.xls 所有样品SNP 统计列表
|---- Genotype.xls 所有样品的基因分型结果
|---- tree 系统发育树结果目录
| |--- nj_tree.out.tre.png 系统发育树分析结果(png格式)
| |--- nj_tree.out.tre.svg 系统发育树分析结果(SVG格式)
|---- PCA PCA分析结果目录
| |--- 1-2.png 第1组分与第2组分贡献率
| |--- 1-3.png 第1组分与第3组分贡献率
| |--- m-n.png 第m组分与第n组分贡献率
|---- structure 群体结构分析结果目录
| |--- STRUCTURE.png 群体结构分析结果(png格式)
| |--- STRUCTURE.svg 群体结构分析结果(SVG格式)
|---- index.html 中文版网页结题报告
|---- index_en.html 英文版网页结题报告